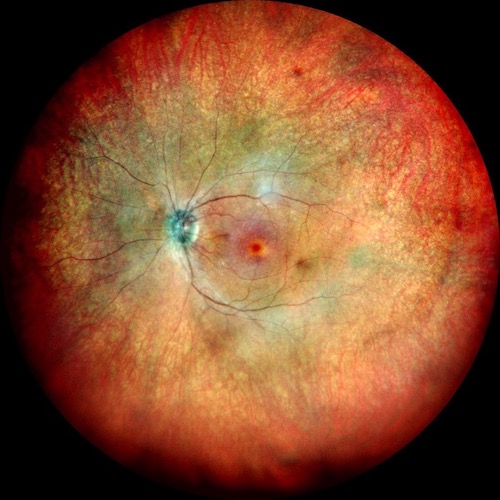
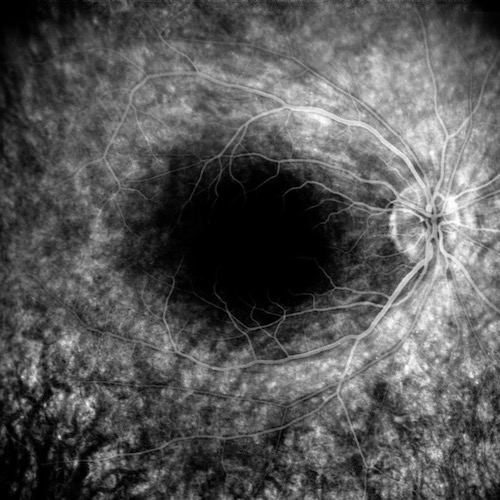
La Retinite Pigmentosa (RP) è una malattia genetica degenerativa della retina, che conduce gradualmente alla perdita della vista. Tra le varie forme che questa patologia può assumere, la Retinite Pigmentosa Sine Pigmento (RPSP) rappresenta una variante atipica particolarmente interessante per la sua peculiarità: l’assenza o la comparsa tardiva delle alterazioni pigmentarie tipiche della RP classica.
La RPSP si distingue per una serie di caratteristiche diagnostiche fondamentali. Primo tra tutti, l’assottigliamento arteriolare, un restringimento dei piccoli vasi sanguigni che irriganano la retina, che porta a un insufficiente apporto di sangue e nutrienti. Il pallore papillare, ovvero un’alterazione del colore della papilla, la regione della retina da cui emerge il nervo ottico, è un altro segnale distintivo. Inoltre, l’elettroretinogramma (ERG), un test che misura l’attività elettrica della retina in risposta alla luce, risulta estinto o marcatamente ridotto nella RPSP, indicando una funzionalità retinica compromessa.
Oltre alla RPSP, esistono altre forme atipiche di RP, come la RP puntata albescente, caratterizzata dalla presenza di piccole macchie bianche diffuse sulla retina; la RP a settore, che coinvolge solamente un quadrante della retina; e la RP pericentrale, che risparmia la retina periferica, concentrandosi sulla zona centrale o vicina al centro.
Queste varianti atipiche di RP possono talvolta essere associate a malattie sistemiche, configurando diverse sindromi. Ad esempio, la Sindrome di Usher combina la RP con la sordità congenita e si manifesta tipicamente prima della pubertà. La Sindrome di Bassen-Kornzweig, caratterizzata da atassia spinocerebellare, assenza di betalipoproteina e malassorbimento dei grassi, è una malattia letale. Altre sindromi che possono presentarsi con la RP includono la Sindrome di Cockayne, con sintomi quali nanismo, invecchiamento precoce e ritardo mentale; la Sindrome di Kearns-Sayre, con oftalmoplegia esterna progressiva e blocco cardiaco; le Mucopolisaccaridosi, con anomalie scheletriche e ritardo mentale; e la Sindrome di Laurence-Moon-Biedl, con ritardo mentale, polidattilia, obesità e ipogonadismo.
La comprensione delle forme atipiche di RP, come la RPSP, è fondamentale non solo per affinare le diagnosi e personalizzare i trattamenti, ma anche per approfondire la conoscenza delle interazioni tra la retina e il resto dell’organismo, aprendo nuove strade nella ricerca di terapie efficaci per queste malattie degenerative della vista.
È giunta alla nostra osservazione un caso di retinite pimentosa sine pigmento: